
Definizione.Storia.Classificazione
Initial posting: 4 Ottobre 2007
last update: Febbraio 2014
Sommario
Definizione
Storia
Classificazione
Materiale didattico
Bibliografia
Sommario
L’Albinismo è un gruppo eterogeneo di anomalie ereditarie della sintesi della melanina, caratterizzato da una riduzione o assenza congenita del pigmento melanico nella cute, nei capelli, nei peli e negli occhi (Albinismo Oculocutaneo, OCA) o quasi esclusivamente negli occhi (Albinismo Oculare, OA). All’ipopigmentazione, cutanea e/o oculare, sono associate determinate anomalie del sistema ottico, in particolare: nistagmo; ipopigmentazione dell'iride, ipopigmentazione della retina e fotofobia; ipoplasia della fovea e riduzione dell’acuità visiva; misrouting ottico e riduzione della visione stereoscopica.
Descrizioni di Albinismo nell’uomo si ritrovano già negli scritti di autori Greci e Latini, come Plinius Secundus il vecchio ed Aulus Gellius. La letteratura medica inizia ben presto a documentare i primi casi di Albinismo, che si rivela essere tra le prime malattie a carattere ereditario.
Le più antiche testimonianze scritte descrivono individui con albinismo come soggetti completamente privi di pigmento visibile: pelle bianca, capelli bianchi e occhi rossi.
L’alta variabilità fenotipica, riscontrata nel corso del tempo, a partire dagli inizi del 900, rende difficile una classificazione clinica dell’Albinismo. Contribuiscono a creare confusione i loci del mantello murino (più di 50 agli inizi degli anni 80).
A partire dagli anni 90 l’analisi molecolare consente di stilare una classificazione più accurata, basata sugli specifici geni coinvolti. Tale classificazione va arricchendosi e meglio delineandosi di anno in anno.
Ad oggi (febbraio 2014), i geni associati con l’Albinismo sono 17/18¤. L’idea comune è che ce ne siano altri, per cui l’attuale classificazione genica è in continua evoluzione.
L’Albinismo viene clinicamente distinto in due forme: l’Albinismo Oculocutaneo e l’Albinismo Oculare, diverse per fenotipo e modello ereditario.
Su base genetica, la prima comprende 17 tipi (OCA1, OCA2, OCA3, OCA4, OCA5, OCA6, OCA7, HPS1-9, CHS), mentre la seconda comprende un solo tipo (OA1).
OCA1-7 e OA1sono asindromici o asistemici; HPS1-9 e CHS sono sindromici o sistemici.
La sindrome di Griscelli non è una forma di Albinismo sindromico, come talvolta indicata, in quanto ad essa non sono associate anomalie del sistema ottico, presenti in tutte le forme di albinismo.
¤Il gene associato ad OCA5 non è stato ancora individuato; di esso si conosce al momento (gennaio 2014) solo la localizzazione cromosomica (chr4q24).
Definizione
L’Albinismo è un gruppo eterogeneo di anomalie ereditarie della sintesi della melanina, caratterizzato da una riduzione o assenza congenita del pigmento melanico nella cute, nei capelli, nei peli e negli occhi (Albinismo Oculocutaneo, OCA) o quasi esclusivamente negli occhi (Albinismo Oculare, OA).
All’ipopigmentazione, cutanea e/o oculare, sono associate determinate anomalie del sistema ottico: ipopigmentazione dell'iride, ipopigmentazione della retina e fotofobia; ipoplasia della fovea e riduzione dell’acuità visiva; riduzione della componente ipsolaterale del tratto ottico e riduzione della visione stereoscopica; nistagmo; strabismo; errori di rifrazione
Storia
Sembra che il termine “albino” sia stato utilizzato per la prima volta da un esploratore Portoghese che, notando, in terre Africane, la presenza di nativi dalla pelle chiara e di nativi dalla pelle scura, pensò, erroneamente, che si trattasse di due razze distinte: chiamò i primi Albini (lat. albus, bianco), i secondi Negri (lat. niger, nero).
Descrizioni di Albinismo nell’uomo si ritrovano, comunque, già negli scritti di autori Greci e Latini, come Plinius Secundus il vecchio ed Aulus Gellius. Di qui la conclusione che l’Albinismo abbia ben presto fatto la sua comparsa nella letteratura medica.
Le più antiche testimonianze scritte descrivono individui con albinismo come soggetti completamente privi di pigmento visibile: pelle bianca, capelli bianchi e occhi rossi.
Le descrizioni si moltiplicano nel corso del tempo, quando si fanno più frequenti i viaggi intorno al mondo, con la conseguente scoperta di nuovi continenti, nuovi popoli, nuovi gruppi etnici.
L’albinismo si colora di superstizioni e pregiudizi, diversi da popolo a popolo, da continente a continente.
Nel corso del XIX secolo, soprattutto in America, gli Albini diventano oggetto di spettacoli ludici, fenomeni da baraccone di circhi ambulanti, soggetti privilegiati da fotografi famosi (Fig. 1):

Fig.1 Famiglia Lucasie.
(Nineteenth Century Images of Albinism)
Classificazione
La “natura familiare” dell’Albinismo viene riconosciuta tanto prima quanto l’Albinismo stesso: è tra le prime malattie ad essere indicata come condizione genetica.
Già agli inizi del 900, vengono a delinearsi due tipi di Albinismo, diversi per fenotipo e per modello ereditario: l’Albinismo oculocutaneo e l’Albinismo oculare.
L’Albinismo oculocutaneo, il classico Albinismo descritto nel corso dei secoli precedenti, coinvolge la pigmentazione cutanea ed oculare ed è autosomico recessivo, presentandosi con uguale probabilità nei maschi e nelle femmine.
L’Albinismo oculare, invece, i cui primi casi vengono individuati all’inizio del secolo, coinvolge solo la pigmentazione oculare ed è X-linked recessivo, presentandosi quasi esclusivamente nei maschi. E’ un professore Inglese, Edward Nettleship, che nel 1909 descrive con molta enfasi le caratteristiche genetiche di un <<…Albinismo oculare incompleto…>>, facendo notare che <<…la discendenza è attraverso la madre in ogni caso; nessun maschio affetto ha un bambino affetto…>>. Tab.1:
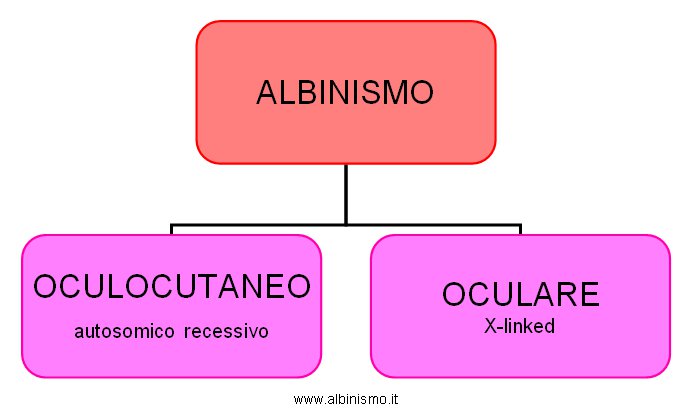
Tab.1 Classificazione dell'Albinismo - inizi 900 -
| Albinismo | - oculocutaneo |
| - oculare |
Ma qual è la natura biochimica dell’Albinismo? Una chiara intuizione viene fornita da Sir Archibald Garrod che si dedica allo studio di alcune malattie ereditarie nell’uomo: alcaptonuria, albinismo, cistinuria, pentosuria. Confrontando la biochimica di individui normali con quella di individui con anomalie ereditarie , pur non conoscendo con precisione le tappe del catabolismo della fenilalanina e della tirosina, ma trovando conferma di quanto supposto nello studio degli alberi genealogici, viene indotto a concludere che certe anomalie ereditarie sono la conseguenza di errori congeniti del metabolismo: <<…un enzima intracellulare è probabilmente assente nei soggetti che presentano queste anomalie…>> (Garrod, 1908). La mancanza di una sufficiente ed attendibile documentazione sperimentale (la dimostrazione biochimica dell’assenza dell’attività della tirosinasi) e la variabilità fenotipica, soprattutto a livello della pigmentazione cutanea, che viene via via evidenziandosi nei soggetti affetti da Albinismo oculocutaneo (si va da soggetti totalmente privi di pigmento a soggetti con una pigmentazione quasi normale), lasciano uno stato di confusione circa la vera natura dell’Albinismo e la sua classificazione. Individui con assenza totale di pigmento, sia cutaneo che oculare, sono facilmente riconoscibili e classificabili. Per essi si parla di Albinismo “completo”, “totale”, “universale” o “perfetto”. Individui che presentano le caratteristiche oculari dell’Albinismo, ma con una qualche pigmentazione cutanea (cioè non una totale assenza di pigmento), identificabili soprattutto nelle popolazioni Africane ed Afro-Americane, creano invece confusione. Per essi si parla di Albinismo “incompleto”, “parziale” o “imperfetto”. Tab. 2:
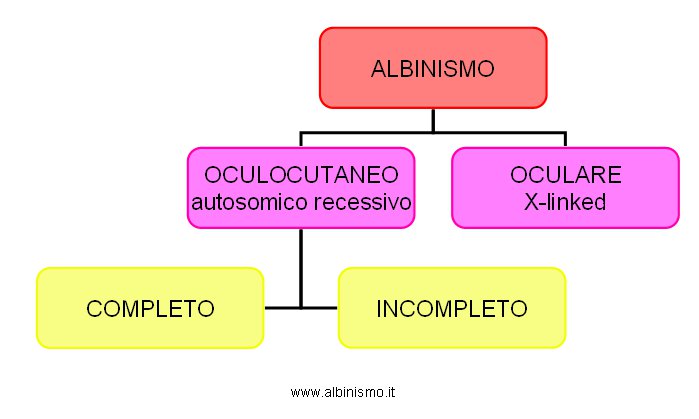
Tab.2 Classificazione delll'Albinismo - inizi 900 -
|
Albinismo |
- oculocutaneo (autosomico recessivo) |
- completo |
|
|
|
- incompleto |
|
|
- oculare (X-linked) |
|
Come spiegare la variabilità fenotipica riscontrata nell’Albinismo oculocutaneo? Gli studi di popolazione indicano che la frequenza di consanguineità in famiglie con albinismo oculocutaneo è più alta di quella attesa nel caso in cui sia coinvolto un solo locus genico, suggerendo così l’eterogeneità genetica per questo disordine.(Stern, 1960, 1977). La prova dell’esistenza di due forme non-alleliche recessive di Albinismo oculocutaneo viene fornita da una famiglia inglese descritta da Trevor-Roper (1952, 1963), in cui entrambi i genitori hanno Albinismo oculocutaneo e la loro progenie presenta una pigmentazione normale (Fig. 2). Questo è il tipico esempio di complementazione genetica classica, dove i genitori sono ognuno omozigote per una mutazione ad un distinto locus e i loro figli pigmentati sono eterozigoti ad entrambi i loci.
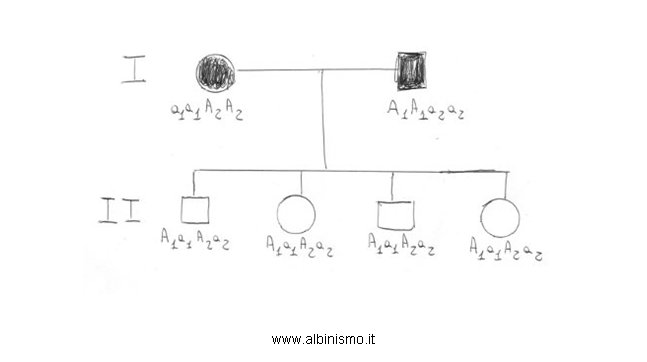
Due genitori inglesi albini hanno quattro figli non albini. Dato che i risultati delle analisi del sangue per la paternità sono compatibili con la legittimità, si può formulare l’ipotesi che i due genitori siano albini per diverse ragioni genetiche, l’ uno perché possiede il genotipo albino a1a1, e l’altro perché ha, in un altro locus, il genotipo albino a2a2. Esprimendo, quindi, i genotipi dei genitori, in una formulazione genetica più elaborata, che preveda l’esistenza di due loci genici, i cui alleli recessivi, in omozigosi, determinano l’albinismo ( l’uno a1a1A2A2, l’altro A1A1a2a2), i figli sarebbero tutti A1a1A2a2 e, dato che A1 è dominante su a1 e A2 è dominante su a2, sarebbero normalmente pigmentati.
E’ probabile che la fratria (generazione II) sia composta da 2 maschi e da 2 femmine (nda.: non è probabilemte corretto l'ordine di nascita)
E’ proprio la presenza di 2 femmine, senza pigmentazione a mosaico del fondo oculare (caratteristica che allora si pensava presente in tutte le femmine eterozigoti per il gene OA1 X-linked), che porta ad escludere, in quegli anni, la possibilità che il padre avesse Albinismo Oculare X-linked e ad avanzare l’ipotesi dell’esistenza di una seconda forma non allelica di Albinismo Oculocutaneo autosomico recessivo.
Da R. Pellegrino, 2010
Witkop (1966, 1970) applicando alla famiglia di Trevor-Roper il test di incubazione del bulbo pilifero, ideato da Kugelman e Van Scott (1961), suggerisce una base biochimica per queste due forme di Albinismo. I bulbi piliferi della madre sono incapaci di produrre melanina quando posti in una soluzione contenente L-tirosina o DOPA (3,4-diidrossifenilalanina), mentre i bulbi piliferi del padre, quando posti nello stesso tipo di soluzione, producono pigmento melanico (Fig. 3). La forma di Albinismo presentata dalla madre viene denominata Albinismo oculocutaneo tirosinasi-negativo (OCA ty-neg, ora OCA1A), mentre quella presentata dal padre viene denominata Albinismo oculocutaneo tirosinasi-positivo (OCA ty-pos, ora OCA2). E’ chiaro che due geni diversi sono responsabili di queste due forme di Albinismo, una non pigmentata e una pigmentata.
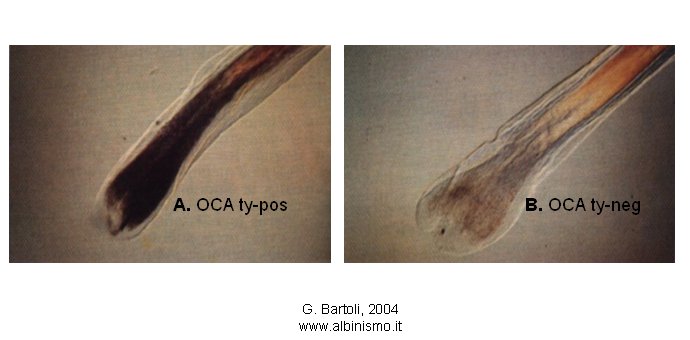
A. OCA ty-pos. B. OCA ty-neg
Fig. 3. Test di incubazione del bulbo pilifero.
I bulbi piliferi vengono incubati per 12-24 ore in una soluzione contenente tirosina o DOPA, a 37°. La sintesi della melanina, in un tubo test, richiede solo la presenza dell’enzima tirosinasi, necessario per le prime due tappe della via biosintetica: l’idrossilazione dell’L-tirosina a 3,4-diidrossifenilalanina (DOPA) e l’ossidazione di DOPA a DOPAchinone. Le reazioni successive procedono spontaneamente. In vivo, invece, intervengono altri enzimi e fattori melanogenici, che determinano il tipo e la quantità di melanina sintetizzata..
A. Il bulbo pilifero diventa scuro. La diagnosi è OCA ty-pos.
B. Il bulbo pilifero rimane chiaro. La diagnosi è OCA ty-neg.
Da G. Bartoli, 2004 - didascalia, Rosa Pellegrino.
In questo modo Witkop dà una prima soluzione al problema della classificazione e della eterogeneità genetica. Tab. 3:
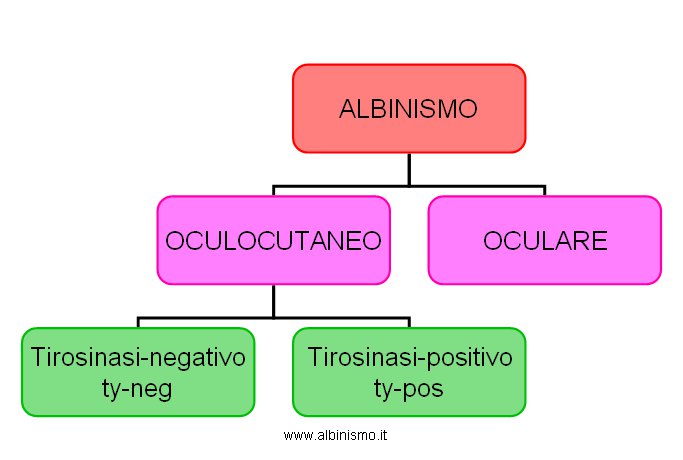
Tab.3 . Classificazione dell'Albinismo - anni 60 -
| Albinismo | - oculocutaneo | - tirosinasi-negativo |
| - tirosinasi-positivo | ||
| - oculare |
La sensibilità del test di incubazione del bulbo pilifero si rivela però, nel corso del tempo, scarsa, dati i falsi positivi e i falsi negativi, legati alla presenza di attività enzimatiche residue.
La scoperta, negli anni 80, di più di 50 loci genici che controllano il colore del mantello nel topo, suggerisce un accurato esame clinico della pigmentazione della cute, dei capelli e degli occhi di individui con Albinismo, atto ad identificare l’equivalente umano di ognuno dei geni murini.
Vengono così identificati diversi “tipi” di Albinismo Oculocutaneo ( OCA tirosinasi-negativo, OCA platino, OCA dal pigmento minimo, OCA giallo, OCA temperatura-sensibile, OCA marrone) e un altro “tipo” di Albinismo oculare ( Albinismo oculare autosomico recessivo, AROA), suggerendo una considerevole eterogeneità di locus, la possibilità quindi che ognuno di questi fenotipi sia causato da un gene diverso. Tab. 4:
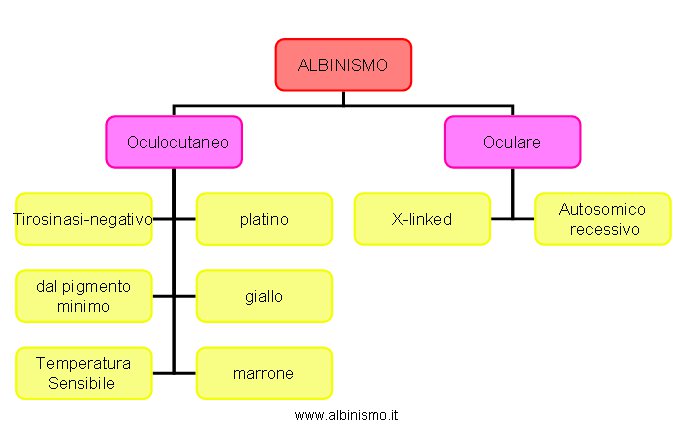
Tab.4 . Classificazione dell' Albinismo - anni 80 -
| Albinismo | - oculocutaneo | - tirosinasi-negativo |
| - platino | ||
| - dal pigmento minimo | ||
| - giallo | ||
| - temperatura sensibile | ||
| - marrone | ||
| -oculare | - X-linked | |
| - autosomico recessivo |
La maggior parte dei geni che influenza il colore del mantello nel topo riguarda lo sviluppo e la distribuzione dei melanociti e il tipo di melanina, piuttosto che la quantità di melanina sintetizzata nel melanocita, e le mutazioni a carico di questi loci non producono albinismo.
A partire dagli anni 90 l’analisi molecolare consente di stilare una classificazione più accurata, basata sugli specifici geni coinvolti (Tab. 5).
Tale classificazione va arricchendosi e meglio delineandosi di anno in anno.
Le correlazioni tra genotipo e fenotipo mostrano che il range fenotipico della pigmentazione per l’Albinismo Oculocutaneo ad ogni locus genico è ampio, comprende molti dei “distinti tipi” di OCA precedentemente descritti in letteratura e può sovrapporsi a quello risultante da mutazioni ad un altro locus genico.
Al 2007, circa 13 sono i geni associati con lo sviluppo dell’Albinismo oculocutaneo e 1 associato con lo sviluppo dell’Albinismo Oculare. L’idea comune è che ce ne siano altri, data l’identificazione di soggetti clinicamente albini, nei quali non sono state trovate mutazioni nei geni noti essere associati all'albinismo (in particolare i più comuni, Tyr e P), almeno nelle sequenze geneticamente esaminabili.
I tipi comuni di Albinismo Oculocutaneo (OCA1, OCA2, OCA3 e OCA4) presentano ipopigmentazione cutanea ed oculare associata con determinati cambiamenti del sistema ottico ( in particolare, ipoplasia foveale e anomale connessioni nervose tra la retina e il cervello), senza significativi coinvolgimenti di altri tessuti. I geni ad essi associati codificano prodotti diversi, ma le mutazioni in tutti, con eterogeneità allelica per ognuno di essi, determinano la riduzione della sintesi della melanina da parte dei melanociti.
I tipi meno comuni di Albinismo Oculocutaneo, come la Sindrome di Hermansky-Pudlak ( HPS1, HPS2, HPS3, HPS4, HPS5, HPS6, HPS7 e HPS8) e la Sindrome di Chediak-Higashi (CHS), presentano manifestazioni fenotipiche più complesse, associate alla presenza di anomalie a carico di vari tipi di organelli specializzati correlati ai lisosomi (LORs).
Tutti i tipi di OCA sono autosomico recessivi.
Il classico OCA tirosinasi-negativo, privo di pigmento, è parte di un ampio spettro di pigmentazione trovato in OCA1 ed è indicato come sottotipo OCA1A.
L’OCA tirosinasi-positivo, pigmentato, può essere causato da mutazioni a diversi loci genici, per cui si parla di diversi tipi di OCA ty-pos: OCA1B ( fenotipi: OCA giallo, OCA dal pigmento minimo, OCA platino, OCA temperatura sensibile), OCA2 (fenotipi: OCA ty-pos classico, OCA marrone), OCA3 (fenotipo OCA rosso), OCA4, HPS e CHS.
L’Albinismo oculare (OA1), presenta ipopigmentazione oculare associata a cambiamenti del sistema ottico, che si ritrovano in tutte le forme di Albinismo, e pigmentazione cutanea dalla riduzione appena percettibile. Il gene ad esso associato, ad eterogeneità allelica, codifica un prodotto che ha un ruolo nella biogenesi dei melanosomi, per cui non varia la quantità di melanina presente, bensì la sua distribuzione.
E’ recessivo X-liked.
L’Albinismo oculare autosomico recessivo (AROA), indicato anche come OA3, rientra in forme leggere di OCA1B e OCA2, dalla pigmentazione quasi normale o normale.
L’ Albinismo Oculare di tipo 2 (OA2), anche noto come Albinismo oculare di tipo Forsius-Eriksson o Disturbo Oculare dell’Isola di Aland (AIED), è una forma allelica della Nictalopia (cecità notturna) incompleta stazionaria congenita (CSNB2A).
Tab. 5:
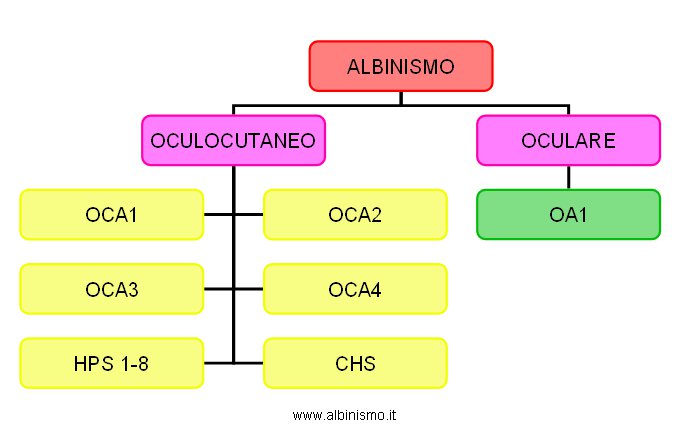
Tab.5. Classificazione dell'Albinismo - 2007 -
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Note.
Sembra confermato che la suddivisione di OCA2 in sottotipi OCA2A e OCA2B non abbia fondamento.
Il gene P viene ora indicato come gene OCA2.
Il gene MATP un tempo veniva indicato come gene AIM-1.
OA1 viene considerato da molti autori come una forma leggera di Albinismo Oculocutaneo (King et altri, 2002 o 2001).
L’ assenza di mutazioni geniche sia nel gene TYR che nel gene P di soggetti clinicamente OCA fa supporre che mutazioni a carico di altri geni determinino l’albinismo.
Da King, 2001 (modificata da Rosa Pellegrino , 2007)
Ad oggi (febbraio 2014), 17 geni sono associati all’Albinismo Oculocutaneo e 1 gene è associato all’Albinismo Oculare.
Circa il 20% dei soggetti clinicamente albini ha una diagnosi genetica irrisolta, in quanto presenta 1 o nessuna mutazione in uno dei geni noti essere associati all'albinismo. Questo fa supporre la presenza di mutazioni in sequenze intergeniche, ma molto più probabilmente l'esistenza di altri geni associati all'albinismo, e quindi altre forme di albinismo.
I geni della pigmentazione sono 378 (al 2011: color genes summary). Poichè solo il 4% circa di essi è stato associato all'albinismo (17 associati ad OCA e 1 ad OA), il campo di indagine è ancora molto ampio, numerosi sono i geni candidati.
Tab. 6
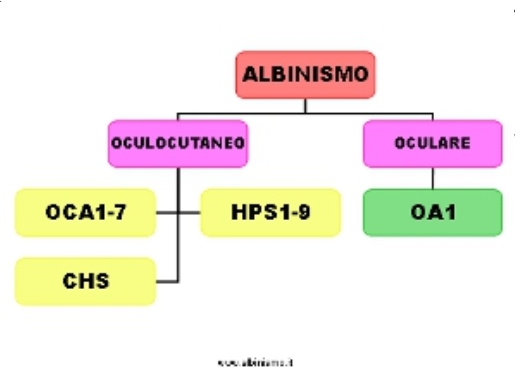
Tab.6. Classificazione dell'Albinismo - 2014 -
| Albinismo. Classificazione genetica - 2014 - | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Note.
n.d. =non definito. Il gene associato ad OCA5 non è stato ancora individuato; di esso si conosce al momento (gennaio 2014) solo la localizzazione cromosomica (chr 4q24)
La sindrome di Griscelli non è una forma di Albinismo sindromico, come talvata indicata, in quanto ad essa non sono associate anomalie del sistema ottico, presenti, in grado variabile, in tutte le forme di albinismo.
Da R. Pellegrino, Quaderno di lezioni sull'albinismo. Appendice 1. 2014
MATERIALE DIDATTICO
Albinismo. Definizione, Storia e Classificazione.pdf
Albinismo. Definizione. Storia.Classificazione. Update 2014.pdf
Storia della Classificazione dell'Albinism.pdf
Albinismo. Classificazione dagli inizi del 900 ad oggi Parte II - 2007/2014. pdf
BIBLIOGRAFIA
Garrod, A. E., Inborn errors of metabolism. Lecture II. Lancet 2, 73-79. 1908.
King, R. A., Hearing, V.G., Creel, D.J. and Oetting, W.S. Albinism. Metabolic and Molecular Bases of Inherited Disease, Ed 8. Scriver, C.R., Beaudet, A.L., Sly, W.S., and Valle, D. (eds). McGraw-Hill, New York; 2001. Text (gentilmente fornito dal prof. R. King).
King, R.A., Summers, C.G., Oetting, W.S., Freyer, J.P., Savage S. International Albinism Center . Facts about Albinism. The clinical Spectrum of Albinism in Human. 2004. Text
Kugelman, T.P., Van Scott, E.J., Tyrosinase activity in melanocytes of human albinos. J. Invest. Dermatol. 37, 73-76. 1961.
Stern, C. Principi di Genetica Umana (pag. 141), Ed. 1. Achtner, W., Battistuzzi, G., Tizzoni, M. Zanichelli, BO. 1977. Text.
Trevor- Roper, P.D. Marriage of two complete albinos with normally pigmented offspring, Br J. Ophthalmol. 36, 107-108. 1952.
Witkop, C.J., Nance, W.E., Rawis, R.F. and White, J.G. Autosomal recessive oculocutaneous albinism in man: Evidence for genetic heterogeneity, Am. J. Hum. Genet. 22, 55-74. 1970.
Pellegrino, R. Quaderno di lezioni sull'albinismo. Appendice 1. 2014
